Státní ústav pro kontrolu léčiv vydává překlad dokumentu Annex 20 k EU GMP, který vstoupil v platnost 1. 3. 2008. Tento dokument vychází z pokynu ICH Q9 Quality risk management a jeho cílem je informovat blíže výrobce léčiv o principech a metodice analýzy rizik, která je součástí výrobních a kontrolních postupů při výrobě léčiv, a tím rovněž obsahem dokumentů předkládaných subjekty regulačním autoritám v rámci registrací.
Předmluva a rozsah působnosti
Nový Doplněk 20 Pokynů SVP odpovídá pokynu ICH Q9 Quality Risk Management (dále QRM, řízení rizik pro jakost). Poskytuje návod pro systematický přístup ke QRM usnadňující dodržování požadavků SVP a dalších požadavků na jakost. Zahrnuje principy, které je třeba uplatnit, a možné procesy, metody a nástroje, které lze použít při uplatňování formálního přístupu k řízení rizik pro jakost.
Z důvodů harmonizace byla provedena revize Pokynů pro SVP, Část I, kapitola 1, Řízení jakosti doplňující aspekty QRM v rámci systému jakosti. Podobná revize se plánuje u Části II Pokynů. Další části pokynů pro SVP mohou být upraveny tak, aby zahrnovaly aspekty QRM, v rámci jejich budoucích významnějších revizí. Revizí kapitol o řízení jakosti v Pokynech pro SVP, Část I a II se QRM stává nedílnou součástí systému jakosti výrobců. Záměrem samotného Doplňku 20 však není vytvářet jakákoliv nová regulační opatření; Doplněk poskytuje přehled mezinárodně uznávaných metod a nástrojů řízení rizik společně se seznamem možného uplatnění podle vlastního uvážení výrobců.Je zřejmé, že pokyn ICH Q9 byl původně vytvořen pro účely QRM v oblasti humánních léčivých přípravků. S implementací v rámci Doplňku 20 jsou přínosy tohoto pokynu, jako jsou postupy, metody a nástroje QRM, dostupné také veterinárnímu sektoru.Zatímco Pokyny pro SVP jsou určeny především pro výrobce, pokyn ICH Q9 se vztahuje i na další pokyny a obsahuje také specifické části pro regulační orgány. Nicméně z důvodů souvislosti a celistvosti byl celý pokyn ICH Q9 převeden do Doplňku 20 Pokynů pro SVP. Další úvahy k aspektům regulace, jako je např. revize „Compilation of Community Procedures on Inspections and Exchange of Information“ a některé další pokyny publikované EMA, budou postupně následovat.
1. Úvod
Principy řízení rizik se účinně uplatňují v mnoha oblastech soukromého a státního sektoru, včetně oblasti financí, pojištění, bezpečnosti práce, veřejného zdraví, farmakovigilance a jsou využívány i orgány regulujícími tyto obory. Přestože v současné době existuje několik příkladů využití QRM ve farmaceutickém průmyslu, jedná se o omezené příklady, které nepředstavují všechny přínosy, které řízení rizik nabízí. Kromě toho ve farmaceutickém průmyslu je uznáván význam systémů jakosti a začíná být zřejmé, že QRM je cennou složkou efektivního systému jakosti. Běžně je přijímána definice rizika jakožto kombinace pravděpodobnosti vzniku škody a závažnosti této škody. Nicméně je obtížné dosáhnout jednotného pohledu různých zainteresovaných stran na aplikaci řízení rizik, neboť každá z nich může vnímat odlišné potencionální škody, vznik různých škod může ohodnotit odlišnou pravděpodobností a odlišným stupněm závažnosti. Ve vztahu k léčivům je třeba považovat ochranu pacienta prostřednictvím řízení rizik pro jakost za věc prvořadého významu, a to i přes velké množství různých zainteresovaných stran, včetně pacientů, lékařů, státu i průmyslu. Výroba a používání léčivého přípravku, včetně jeho složek, nezbytně zahrnuje jistý stupeň rizika. Riziko jeho jakosti je pouze jednou složkou celkového rizika. Je důležité chápat, že jakost přípravku je třeba zachovat po celou dobu životního cyklu přípravku tak, aby vlastnosti, které jsou důležité pro jakost léčivého přípravku, stále odpovídaly těm, které byly použity v klinických studiích. Efektivní použití QRM může dále zajistit vysokou kvalitu léčivého přípravku pro pacienta tím, že poskytne proaktivní prostředky pro stanovení a kontrolu potencionálních problémů jakosti během vývoje a výroby. Kromě toho může využití QRM zlepšit rozhodování v případě, že vyvstane problém s jakostí. Efektivní QRM může napomoci lepšímu a informovanějšímu rozhodování, může regulační autority lépe ujistit o schopnosti společnosti vypořádat se s potencionálními riziky a může mít příznivý vliv na rozsah a úroveň regulačního dohledu. Účelem tohoto dokumentu je nabídnout systematický přístup ke QRM. Dokument slouží jako základní dokument či podklad, který je nezávislý na ostatních dokumentech jakosti ICH, nicméně je podporuje a doplňuje stávající praxi, požadavky, standardy a pokyny v oblasti jakosti v rámci farmaceutického průmyslu a regulačního prostředí. Konkrétně poskytuje pokyny k principům a některým nástrojům QRM, které mohou umožnit efektivnější a konzistentnější rozhodování na základě rizik, a to jak ze strany regulačních autorit, tak průmyslu, ve smyslu jakosti léčivých látek a léčivých přípravků po celou dobu životního cyklu přípravku. Záměrem dokumentu není vytvářet další nová očekávání nad rámec stávajících regulačních požadavků.Není vždy vhodné ani nezbytné uplatňovat formální postup řízení rizik (s využitím uznávaných nástrojů a/nebo interních postupů, např. standardních operačních postupů). Použití neformálních postupů řízení rizik (použití empirických nástrojů a/nebo interních postupů) lze také považovat za přijatelné. Vhodné použití QRM může pomoci při plnění povinnosti průmyslu dodržovat regulační požadavky, nicméně této povinnosti nezbavuje ani nenahrazuje vhodnou komunikaci mezi průmyslem a regulačními autoritami.
2. Rozsah
Tento pokyn uvádí principy a příklady nástrojů QRM, které lze aplikovat na různé aspekty farmaceutické jakosti. Tyto aspekty zahrnují vývoj, výrobu, distribuci a inspekci a dále postupy předkládání/kontroly po dobu životního cyklu léčivých látek, léčivých přípravků, biologických a biotechnologických přípravků (včetně použití surovin, rozpouštědel, pomocných látek, obalových materiálů a materiálů pro značení léčivých přípravků a biologických a biotechnologických přípravků).
3. Principy QRM
Dva základní principy QRM jsou tyto:
- vyhodnocení rizika pro jakost má být založeno na vědeckých znalostech a v konečném důsledku spojeno s ochranou pacienta; a
- pracnost, formálnost a dokumentace procesu QRM by měly být úměrné úrovni rizika.
4. Obecný proces QRM
QRM představuje systematický proces posouzení, kontroly, sdělování a přehodnocení rizik pro jakost léčivého přípravku po celou dobu životního cyklu přípravku. Model QRM je zobrazen v diagramu (obr.1). Využít lze i jiné modely. Důraz na každou složku struktury se může lišit případ od případu, ale robustní proces bude zahrnovat zvážení všech prvků v takových podrobnostech, jaké odpovídají konkrétnímu riziku.
Obr. 1
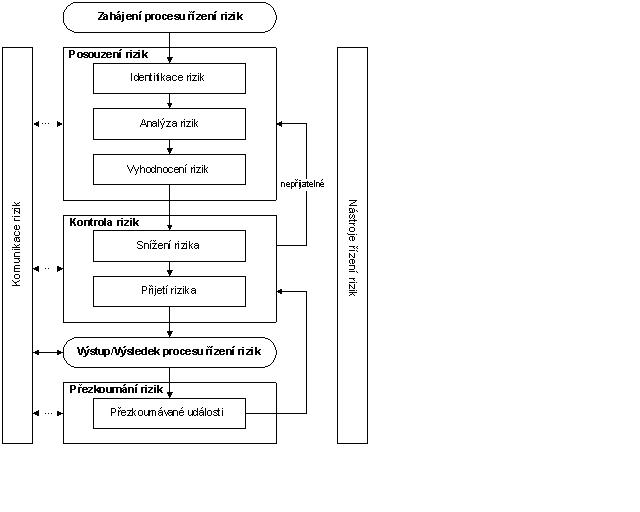 Rozhodovací kroky nejsou na výše uvedeném diagramu znázorněny, protože k rozhodnutí může dojít kdykoliv během QRM procesu. Takovým rozhodnutím může být rozhodnutí o návratu k předchozímu kroku a zjištění dalších informací, úpravě modelů rizik a dokonce o ukončení procesu řízení rizik na základě informací, které takovéto rozhodnutí podporují.
Rozhodovací kroky nejsou na výše uvedeném diagramu znázorněny, protože k rozhodnutí může dojít kdykoliv během QRM procesu. Takovým rozhodnutím může být rozhodnutí o návratu k předchozímu kroku a zjištění dalších informací, úpravě modelů rizik a dokonce o ukončení procesu řízení rizik na základě informací, které takovéto rozhodnutí podporují.
Pozn.: „Nepřijatelné" ve vývojovém diagramu se netýká pouze zákonných, legislativních či regulačních požadavků, ale také potřeby znovu přezkoumat proces posouzení rizik.
4.1 Odpovědnosti
Činnosti QRM obvykle, nikoliv však vždy, provádí interdisciplinární týmy. Týmy by měly vedle osob znalých procesu QRM zahrnout také odborníky z příslušných oblastí (např. útvar jakosti, obchodní rozvoj, technický útvar, registrace, výroba, prodej a marketing, oblast právní, statistická a klinická).
Pracovníci, kteří rozhodují, mají:
- převzít odpovědnost za koordinaci QRM napříč různými funkcemi a odděleními své organizace, a
- zajistit, aby proces QRM byl definován, rozvíjen a přezkoumáván a aby byly kdispozici odpovídající zdroje.
4.2 Zavedení procesu QRM
QRM by měl zahrnovat systematické postupy navržené za účelem koordinace, podpory a zlepšování vědeckého rozhodování o rizicích. Možné kroky učiněné v zavádění a plánování procesu QRM mohou zahrnovat následující:
- definování problému a/nebo otázky rizika, včetně příslušných předpokladů určujících potenciál rizika,
- shromáždění podkladů a/nebo dat o potenciálním nebezpečí, škodě či dopadu na zdraví lidí vsouvislosti sposouzením rizika,
- určení vedoucího pracovníka a nezbytných zdrojů,
- stanovení termínů a úkolů a vhodné úrovně rozhodování pro proces řízení rizik.
4.3 Posouzení rizik
Posouzení rizik sestává z určení nebezpečí a analýzy a vyhodnocení rizik souvisejících s vystavením tomuto nebezpečí (jak je definováno níže).
Posouzení rizik začíná správně stanoveným popisem problému nebo otázkou rizika. Pokud je dané riziko správně definováno, bude snadnější určit vhodný nástroj řízení rizik (viz příklady v části 5) a druh informací potřebných k řešení otázky rizika. Jako pomůcka pro jasnou definici rizik(a) pro účely posouzení rizik často poslouží tyto tři základní otázky:
- Co by mohlo selhat?
- Jaká je pravděpodobnost tohoto selhání?
- Jaké jsou důsledky (závažnost)?
Identifikace rizik představuje systematické využití informací pro stanovení nebezpečí vztahujícímu se k riziku nebo popisu problému. Informace mohou obsahovat historická data, teoretickou analýzu, informovaná stanoviska a obavy a zájmy zainteresovaných stran. Identifikace rizika odpovídá na otázku „Co by mohlo selhat?", a to včetně určení možných následků. Tak se získá východisko pro další kroky v procesu QRM.
Analýza rizik představuje odhad rizik souvisejících s určenými nebezpečími. Jedná se o kvalitativní či kvantitativní proces stanovení vazeb mezi pravděpodobností výskytu a závažností škod. U některých nástrojů řízení rizik je jedním z faktorů odhadu rizika také schopnost zjistit škodu (detekovatelnost).
Vyhodnocení rizik porovnává stanovené a analyzované riziko s danými rizikovými kritérii. Vyhodnocení rizika zvažuje pádnost důkazů u všech tří základních otázek.
Při efektivním posuzování rizik je významná robustnost datového souboru, neboť je rozhodující pro kvalitu výstupu. Odhalující předpoklady a racionální zdroje nejistoty zvýší důvěryhodnost tohoto výstupu a/nebo napomůže určit jeho omezení. Nejistota je dána kombinací neúplných znalostí procesu a jeho předpokládané či nepředpokládané variability. Typickými zdroji nejistoty jsou mezery ve znalostech farmacie a chápání procesu, zdrojů škod (např. způsoby selhání v procesu, zdroje variability) a pravděpodobnost detekce problémů.
Výstupem z posouzení rizik je buď kvantitativní odhad rizika nebo kvalitativní popis rozsahu rizika. Pokud je riziko vyjádřeno kvantitativně, použije se numerická pravděpodobnost. Jinak lze riziko vyjádřit pomocí kvalitativních deskriptorů, jako je „vysoké", „střední" nebo „nízké", které je však třeba co nejpodrobněji definovat. Někdy se pro podrobnější vymezení deskriptorů v hodnocení rizika použije „skóre rizika". Při kvantitativním posouzení rizika uvádí odhad rizika pravděpodobnost konkrétního důsledku, a to na základě daného souboru okolností, za nichž může riziko vyvstat. Kvantitativní odhad rizika je tedy užitečný vždy pro jeden konkrétní důsledek. Jinak některé nástroje řízení rizik používají relativní měřítko rizika, kdy se kombinují různé úrovně závažnosti a pravděpodobnosti, a tak se vytváří celkový odhad relativního rizika. Mezikroky v procesu hodnocení mohou někdy využívat kvantitativního odhadu rizika.
4.4 Kontrola rizik
Kontrola rizik zahrnuje rozhodování o snížení a/nebo přijetí rizik. Účelem kontroly rizik je snížit riziko na přijatelnou úroveň. Úsilí vynaložené na kontrolu rizik má být úměrné významu rizika. Pracovníci, kteří rozhodují, mohou pro pochopení optimální úrovně kontroly rizik využít různé postupy, včetně analýzy prospěšnosti a nákladů.
Kontrola rizik by se mohla zaměřovat na následující otázky:
- Přesahuje riziko přijatelnou úroveň?
- Co lze učinit ke snížení nebo odstranění rizik?
- Jaká je vhodná rovnováha mezi prospěchem, riziky a zdroji?
- Vznikají vdůsledku kontroly identifikovaných rizik nějaká nová rizika?
Snížení rizika se zaměřuje na procesy zmírnění nebo prevence vzniku rizika pro jakost, pokud toto překročí určenou (přijatelnou) úroveň (viz obr. 1). Snížení rizika může zahrnovat opatření vedoucí ke snížení závažnosti a pravděpodobnosti škody. V rámci strategie kontroly rizik lze také využít procesy, které zlepšují detekovatelnost nebezpečí a rizik pro jakost. Implementace opatření směřujících ke zmírnění rizika může do systému vnést nová rizika nebo zvýšit význam jiných stávajících rizik. Proto může být vhodné revidovat posouzení rizik po zavedení procesu snižování rizik, aby se určily a vyhodnotily jakékoliv případné změny v rizicích.
Přijetí rizika představuje rozhodnutí o tom, že riziko bude přijato. Přijetí rizika může mít podobu formálního rozhodnutí o přijetí zbytkového rizika nebo se může jednat o pasivní rozhodnutí, v němž nejsou zbytková rizika specifikována. U některých druhů škod dokonce ani nejlepší praxe QRM nemůže zcela vyloučit riziko. Za takovýchto okolností se lze shodnout, že byla uplatněna odpovídající strategie QRM a že riziko jakosti je sníženo na určenou (přijatelnou) úroveň. Tato (specifikovaná) přijatelná úroveň bude záviset na mnoha parametrech a je třeba o ní rozhodovat individuálně.
4.5 Komunikace rizik
Komunikace rizik je sdílení informací o riziku a řízení rizik mezi pracovníky, kteří rozhodují, a ostatními. Strany spolu mohou komunikovat v jakékoliv fázi procesu řízení rizik (viz obr. 1 - čárkované šipky). Výstup/výsledek procesu QRM by měl být vhodně sdělen a zdokumentován (viz obr. 1 - plná šipka). Komunikace může být vedena mezi zainteresovanými stranami, např. regulačními autoritami a průmyslem, průmyslem a pacientem, v rámci podniku, průmyslu či regulačního orgánu apod. Sdělované informace se mohou vztahovat k existenci, povaze, formě, pravděpodobnosti, závažnosti, přijatelnosti, kontrole, zacházení, detekovatelnosti či jiným aspektům rizika pro jakost. Komunikace se nemusí uskutečňovat ohledně přijetí každého rizika. Komunikace o rozhodnutích QRM mezi průmyslem a regulačními orgány může probíhat prostřednictvím stávajících kanálů, jak je uvedeno v předpisech a pokynech.
4.6 Přezkoumání rizik
Řízení rizik by mělo být trvalou součástí procesu řízení jakosti. Je třeba zavést mechanismus pro přezkoumání nebo monitorování událostí. Výstupy/výsledky procesu řízení rizik mají být přezkoumávány, aby byly zohledněny nové poznatky a zkušenosti. Jakmile se zavede proces QRM, měl by se i nadále používat pro události, které by mohly mít vliv na původní rozhodnutí QRM, a to bez ohledu na to, zda se jedná o plánované události (např. výsledky přehodnocení přípravku, inspekce, audity, kontrola změn) nebo neplánované příhody (např. původní příčina z šetření selhání, stažení). Frekvence přezkoumání by měla vycházet z úrovně rizika. Přezkoumání rizika může zahrnovat přehodnocení rozhodnutí o přijetí rizika (část 4.4.).
5. Metodika řízení rizik
QRM podporuje vědecký a praktický přístup k rozhodování. Poskytuje dokumentované, transparentní a reprodukovatelné metody, jak dosáhnout kroků v procesu QRM na základě aktuálních znalostí o posuzování pravděpodobnosti, závažnosti a někdy detekovatelnosti rizika.
Rizika pro jakost byla tradičně posuzována a řízena nejrůznějšími neformálními způsoby (empirickými a/nebo interními postupy), založenými např. na kompilaci pozorování, trendů a dalších informací. Tyto přístupy i nadále poskytují užitečné informace, které mohou podpořit např. řešení stížností, závad v jakosti, odchylek a alokace zdrojů.
Kromě toho mohou regulační autority a farmaceutický průmysl posuzovat a řídit rizika pomocí uznávaných nástrojů a/nebo interních postupů (např. standardních operačních postupů) pro řízení rizik. Následuje neúplný seznam některých z těchto nástrojů (další podrobnosti jsou uvedeny v Příloze 1 a v kapitole 8):
- Základní metody usnadňující řízení rizik (vývojové diagramy, kontrolní listy apod.)
- Analýza možných vad a jejich důsledků (FMEA, Failure Mode Effects Analysis)
- Analýza možných vad, jejich důsledků a kritičnosti (FMECA, Failure Mode, Effects and Criticality Analysis)
- Analýza stromu poruchových stavů (FTA, Fault Tree Analysis)
- Analýza nebezpečí a kritické kontrolní body (HACCP, Hazard Analysis and Critical Control Points)
- Analýza ohrožení a provozuschopnosti (HAZOP, Hazard Operability Analysis)
- Předběžná analýza nebezpečí (PHA, Preliminary Hazard Analysis)
- Klasifikace a filtrování rizik
- Podpůrné statistické nástroje
Může být vhodné uplatnit tyto nástroje v konkrétních oblastech týkajících se jakosti léčivých látek a léčivých přípravků. Metody QRM a podpůrné statistické nástroje lze kombinovat (např. pravděpodobnostní odhad rizik). Kombinované použití nabízí flexibilitu, která může usnadnit aplikaci principů QRM.
Stupeň přísnosti a formálnosti aplikace QRM by měl odrážet dostupné znalosti a měl by odpovídat složitosti a/nebo kritičnosti řešeného problému.
6. Integrace QRM do činností průmyslu a regulačních autorit
QRM je proces, který podporuje vědecké a praktické rozhodování, když je začleněn do systémů jakosti (viz Příloha II). Jak bylo nastíněno v Úvodu, vhodné uplatnění QRM nezbavuje průmysl povinnosti plnit regulační požadavky. Nicméně efektivní QRM může usnadnit lepší a informovanější rozhodování, může pro regulátory znamenat větší záruku toho, že společnost je schopna vypořádat se s potencionálními riziky, a může ovlivnit rozsah a úroveň přímého regulačního dohledu. Kromě toho může QRM podpořit lepší využívání zdrojů všemi stranami.
Vyškolení pracovníků průmyslu i regulačních orgánů v procesech QRM zajišťuje lepší pochopení postupů rozhodování a zvyšuje důvěryhodnost výsledků QRM.
QRM by mělo být vhodným způsobem začleněno do stávajících činností a zdokumentováno. Příloha II uvádí příklady situací, v nichž uplatnění procesu QRM může poskytnout informace, které lze využít v řadě různých činností v oblasti farmacie. Tyto příklady slouží pouze pro ilustraci a jejich výčet nelze považovat za definitivní či vyčerpávající. Cílem těchto příkladů není vytvořit žádná nová očekávání nad rámec požadavků stanovených stávajícími předpisy.
Příklady pro činnosti průmyslu a regulace (viz Příloha II):
- Řízení jakosti
Příklady pro provozní a další činnosti průmyslu (viz Příloha II):
- Vývoj
- Vybavení, zařízení a inženýrské sítě
- Skladové hospodářství
- Vlastní výroba
- Laboratorní kontrola a zkoušky stability
- Balení a značení
Příklady pro regulační činnosti (viz Příloha II):
- Činnosti inspekce a posuzování
Přestože regulační rozhodnutí budou i nadále přijímána regionálně, jednotné chápání a uplatňování principů QRM by mohlo zvýšit vzájemnou důvěru a podpořit jednotnější rozhodování regulátorů vycházející ze stejných informací. Tato spolupráce by mohla být významná při tvorbě politik a pokynů integrujících a podporujících praxe QRM.¨
7. Definice
Pracovník, který rozhoduje (Decision maker) - osoba kompetentní a oprávněná k tomu, aby mohla vhodně a včas rozhodovat v otázkách QRM.
Detekovatelnost (Detectability) - schopnost odhalit nebo stanovit existenci, přítomnost nebo skutečnost nebezpečí.
Škoda (Harm) - újma na zdraví, včetně poškození, k němuž může dojít v důsledku ztráty kvality či dostupnosti přípravku.
Nebezpečí (Hazard) - potencionální zdroj škody (ISO/IEC Pokyn 51)
Životní cyklus přípravku (Product Lifecycle) - všechny fáze existence přípravku, od počátečního vývoje, přes uvedení na trh až po ukončení jeho výroby
Jakost (Quality) - míra, s níž soubor inherentních vlastností přípravku, systému nebo procesu splňuje požadavky (viz definici ICH Q6a konkrétně pro „jakost" léčivé látky a léčivých přípravků).
QRM, řízení rizik pro jakost (Quality risk management) - systematický proces posuzování, kontroly, komunikace a přezkoumávání rizik pro jakost léčivého přípravku po celou dobu jeho životního cyklu.
Systém jakosti (Quality system) - souhrn všech aspektů systému, který implementuje politiku jakosti a zajišťuje plnění cílů jakosti.
Požadavky (Requirements) - explicitní nebo implicitní potřeby či očekávání pacientů nebo osob, které je zastupují (např. zdravotníků, regulátorů a zákonodárců). V tomto dokumentu „požadavky" zahrnují nejenom zákonné, legislativní či regulační požadavky, ale také takovéto potřeby a očekávání.
Riziko (Risk) - kombinace pravděpodobnosti vzniku škody a závažnosti této škody (ISO/IEC Pokyn 51).
Přijetí rizika (Risk acceptance) - rozhodnutí o přijetí rizika (ISO Pokyn 73).
Analýza rizik (Risk analysis) - odhad rizika spjatého se stanoveným nebezpečím.
Posouzení rizika (Risk assessment) - systematický proces uspořádání informací takovým způsobem, aby podporovaly rozhodování o riziku, které má být učiněno v rámci procesu řízení rizik. Sestává z určení nebezpečí a analýzy a vyhodnocení rizik souvisejících s vystavením tomuto nebezpečí.
Komunikace rizik (Risk communication) - sdílení informací o riziku a řízení rizik mezi pracovníkem, který rozhoduje, a dalšími zainteresovanými stranami.
Kontrola rizika (Risk control) - kroky, jimiž se realizují rozhodnutí o řízení rizik (ISO Pokyn 73)
Vyhodnocení rizika (Risk evaluation) - porovnání odhadovaného rizika s danými kritérii rizika pomocí kvantitativní a kvalitativní škály s cílem stanovit význam rizika.
Identifikace rizika (Risk identification) - systematické využívání informací pro stanovení potencionálních zdrojů škod (nebezpečí) souvisejících s otázkou rizika nebo popisem problému.
Řízení rizik (Risk management) - systematické uplatňování politik, postupů a praktik systému jakosti pro posuzování, kontrolu, komunikaci a přezkoumávání rizika.
Snížení rizika (Risk reduction) - kroky podniknuté ke snížení pravděpodobnosti vzniku škody a závažnosti této škody.
Přezkoumání rizika (Risk review) - přezkoumání nebo sledování výstupů/výsledků procesu řízení rizik, přičemž se (je-li to vhodné) uplatňují nové poznatky a zkušenosti týkající se daného rizika.
Závažnost (Severity) - měřítko možných důsledků nebezpečí.
Zainteresovaná strana (Stakeholder) - jakákoliv fyzická osoba, skupina nebo organizace, která může ovlivnit riziko nebo být ovlivněna či cítit se ovlivněna rizikem. Pracovníci, kteří rozhodují, mohou být zároveň zainteresovanými stranami. Pro účely tohoto pokynu jsou primárními zainteresovanými stranami pacient, zdravotník, regulační orgán a průmysl.
Trend (Trend) - statistický termín vyjadřující směr nebo rychlost změny proměnné (proměnných).
8. Odkazy
- ICH Q8 Pharmaceutical development
- ISO/IEC Guide 73:2002 - Risk Management - Vocabulary - Guidelines for use in
- Standards
- ISO/IEC Guide 51:1999 - Safety Aspects - Guideline for their inclusion in standards
- Process Mapping by the American Productivity & Quality Center 2002, ISBN
- 1928593739
- IEC 61025 - Fault Tree Analysis (FTA)
- IEC 60812 Analysis Techniques for system reliability-Procedures for failure mode
- and effects analysis (FMEA)
- Failure Mode and Effect Analysis, FMEA from Theory to Execution, 2nd Edition
- 2003, D. H. Stamatis, ISBN 0873895983
- Guidelines for Failure Modes and Effects Analysis (FMEA) for Medical Devices,
- 2003 Dyadem Press ISBN 0849319102
- The Basics of FMEA, Robin McDermott, Raymond J. Mikulak, Michael R.
- Beauregard 1996 ISBN 0527763209
- WHO Technical Report Series No 908, 2003 Annex 7 Application of Hazard Analysis
- and Critical Control Point (HACCP) methodology to pharmaceuticals.
- IEC 61882 - Hazard Operability Analysis (HAZOP)
- ISO 14971:2000 - Application of Risk Management to Medical Devices
- ISO 7870:1993 - Control Charts
- ISO 7871:1997 - Cumulative Sum Charts
- ISO 7966:1993 - Acceptance Control Charts
- ISO 8258:1991 - Shewhart Control Charts
- What is Total Quality Control; The Japanese Way, Kaoru Ishikawa (Translated by
- David J. Liu, 1985, ISBN 0139524339
Příloha I: Metody a nástroje řízení rizik
Účelem této přílohy je poskytnout obecný přehled a odkazy na některé základní nástroje, které mohou použít regulátoři a průmysl v QRM. Odkazy jsou začleněny jako pomůcka pro získání dalších poznatků a podrobností o konkrétním nástroji. Nejedná se o vyčerpávající seznam. Je důležité mít na paměti, že žádný nástroj či soubor nástrojů nelze využít ve všech situacích, kdy se uplatňuje QRM.
I.1 Základní metody usnadňující řízení rizik
Některé z jednoduchých technik, které se běžně používají pro strukturování řízení rizik pomocí uspořádání dat a usnadnění rozhodování, jsou tyto:
- Vývojové diagramy
- Kontrolní listy
- Mapování procesů
- Diagram příčin a následků (jinak též Ishikawův diagram či diagram typu „ rybí kost")
I.2 Analýza FMEA
FMEA (viz IEC 60812) umožňuje vyhodnocení možných způsobů selhání procesů a jejich pravděpodobný dopad na výsledky a/nebo vlastnosti produktu. Jakmile se možné způsoby selhání stanoví, lze použít snížení rizika pro eliminaci, zachování, snížení či kontrolu potencionálních selhání. Analýza FMEA je založena na znalostech produktu a procesu. Metodicky rozebírá analýzu komplexních procesů na zvladatelné kroky. Jedná se o silný nástroj pro shrnutí důležitých způsobů selhání, faktorů způsobujících tato selhání a pravděpodobných dopadů těchto selhání.
Možné oblasti využití
Analýzu FMEA lze využít pro stanovení priorit rizik a sledování účinnosti kontroly rizik.
Lze ji uplatnit u zařízení a vybavení a použít ji pro analýzu výrobní činnosti a jejího vlivu na produkt nebo proces. Identifikuje prvky/činnosti v rámci systému, které zvyšují jeho zranitelnost. Výstup/výsledek analýzy FMEA je možno využít jako podklad pro koncepci nebo další analýzu či jako vodítko pro nasazení zdrojů.
I.3 Analýza FMECA
Analýzu FMEA lze rozšířit tak, aby zahrnula šetření stupně závažnosti důsledků, pravděpodobnost jejich vzniku a jejich detekovatelnost - tak vzniká analýza možných vad, jejich důsledků a kritičnosti (FMECA; viz IEC 60812). K tomu, aby bylo možno takovouto analýzu uskutečnit, je třeba stanovit specifikace produktu nebo procesu.
FMECA může určit místa, kde by mohlo být vhodné přijmout doplňující preventivní opatření, aby se minimalizovala rizika.
Možné oblasti využití
Uplatnění analýzy FMECA ve farmaceutickém průmyslu se předpokládá především u selhání a rizik souvisejících s výrobním postupem; nicméně neomezuje se pouze na toto použití. Výstupem analýzy FMECA je relativní „skóre" rizik pro jednotlivé způsoby selhání, které se používá pro klasifikaci těchto způsobů podle relativních rizik.
I.4 Analýza FTA
Nástroj FTA (viz IEC 61025) představuje přístup, který předpokládá selhání funkčnosti produktu nebo procesu. Tento nástroj vyhodnocuje selhání systému (nebo subsystému) jedno po druhém, přičemž může kombinovat více příčin selhání tak, že určí kauzální řetězce. Výsledky se znázorňují obrazově v podobě stromu různých chyb. Na každé úrovni stromu jsou popsány kombinace různých chyb logickými operátory (A, NEBO apod.). Analýza FTA je založena na odborném pochopení procesu, z nějž vychází identifikace kauzálních faktorů.
Možné oblasti využití
Analýzu FTA lze využít k stanovení cesty k původní příčině selhání. Lze ji uplatnit při šetření stížností nebo odchylek, kdy umožňuje úplné pochopení jejich původních příčin a zajišťuje, že zamýšlené zlepšení plně vyřeší problém a nepovede ke vzniku dalších problémů (tj. vyřeší jeden problém, ale vyvolá další). Analýza FTA představuje účinný nástroj pro vyhodnocení toho, jak několik různých faktorů ovlivňuje daný problém. Výstup analýzy FTA zahrnuje vizuální znázornění způsobů selhání. Je užitečný jak pro posouzení rizik, tak pro vývoj monitorovacích programů.
I.5 Analýza HACCP
Analýza HACCP představuje systematický, proaktivní a preventivní nástroj pro zajištění jakosti, spolehlivosti a bezpečnosti produktu (viz WHO Technical Report Series No 908, 2003 Annex 7). Jedná se o strukturovaný přístup, který uplatňuje technické a vědecké principy s cílem analýzy, vyhodnocení, prevence a kontroly rizika či nežádoucích důsledků nebezpečí daných návrhem, vývojem, výrobou a používáním produktů.
Analýza HACCP sestává z následujících sedmi kroků:
(1) uskutečnění analýzy nebezpečí a identifikace preventivních opatření pro každý krok procesu,
(2) stanovení kritických kontrolních bodů,
(3) stanovení kritických limitů,
(4) zavedení systému sledování kritických kontrolních bodů,
(5) stanovení nápravného opatření, které je třeba přijmout, pokud sledování ukazuje, že kritické kontrolní body nejsou pod kontrolou,
(6) zavedení systému ověřování účinnosti systému HACCP,
(7) zavedení systému uchovávání záznamů.
Možné oblasti využití
Analýzu HACCP lze využít pro stanovení a řízení rizik souvisejících s fyzikálními, chemickými či biologickými nebezpečími (včetně mikrobiologické kontaminace). Analýza HACCP je nejužitečnější, pokud je znalost produktu a procesu dostatečně komplexní, aby podporovala identifikaci kritických kontrolních bodů. Výstupem analýzy HACCP je informace pro řízení rizik, která usnadňuje sledování kritických bodů nejenom ve výrobním procesu, ale také v jiných fázích životního cyklu.
I.6 Analýza HAZOP
Analýza HAZOP (viz IEC 61882) vychází z teorie, která předpokládá, že rizikové události jsou způsobeny odchylkami od projektu či provozních záměrů. Jedná se o systematickou techniku brainstormingu, jíž se identifikují nebezpečí pomocí tzv. klíčových slov. Tato klíčová slova („guide-words", např. Ne/Žádný, Více, Jiný než, Část apod.) se aplikují na příslušné parametry (např. kontaminace, teplota), a tak napomáhají stanovit potencionální odchylky od běžného použití nebo projektových záměrů. Často se využívá týmu osob s odbornými zkušenostmi zahrnujícími navrhování procesu nebo produktu a jejich použití.
Možné oblasti využití
Analýzu HAZOP lze použít na výrobní postupy, včetně kontrahované výroby a formulace i na dodavatelské vztahy, zařízení a vybavení pro léčivé látky a léčivé přípravky. Ve farmaceutickém průmyslu se používá především pro vyhodnocování bezpečnostních rizik procesů. Podobně jako u analýzy HACCP je výstupem analýzy HAZOP seznam činností kritických z hlediska řízení rizik. To napomáhá pravidelnému monitorivání kritických bodů ve výrobním procesu.
I.7 Analýza PHA
Analýza PHA představuje analytický nástroj založený na aplikaci předchozích zkušeností či znalostí nebezpečí či selhání pro účely identifikace budoucích nebezpečí, nebezpečných situací a událostí, které by mohly způsobit škody, a dále pro odhad pravděpodobnosti jejich výskytu v dané činnosti, zařízení, produktu či systému. Tento nástroj sestává z: 1) identifikace možností výskytu rizikové události, 2) kvalitativního vyhodnocení rozsahu možné újmy či poškození zdraví, k němuž by následně mohlo dojít, a 3) relativní klasifikace nebezpečí vycházející z kombinace závažnosti a pravděpodobnosti výskytu a 4) identifikace možných nápravných opatření.
Možné oblasti využití
Analýza PHA může být užitečná při rozboru stávajících systémů či stanovení priorit nebezpečí tam, kde okolnosti neumožňují uplatnění komplexnější metody. Lze ji využít u navrhování produktu, procesu či zařízení, i pro hodnocení typů nebezpečí u obecného typu produktu, poté třídy produktu a konečně konkrétního produktu. Analýza PHA se velmi často používá v raných fázích vývoje projektu, kdy je k dispozici jen málo informací o podrobnostech projektu či provozních postupech; je tedy často předchůdcem dalších studií.
Nebezpečí určená analýzou PHA se obvykle dále posuzují pomocí dalších nástrojů pro řízení rizik, jako jsou ty, které zmiňuje tato část.
I.8 Klasifikace a filtrování rizik
Klasifikace a filtrování rizik je nástroj pro srovnání a klasifikaci rizik. Klasifikace rizik v komplexních systémech obvykle vyžaduje vyhodnocení mnoha různých kvantitativních a kvalitativních faktorů jednotlivých rizik. Tento nástroj zahrnuje rozložení základní otázky rizika do tolika složek, kolik je potřeba k podchycení faktorů obsažených v daném riziku. Tyto faktory se kombinují do jediného skóre relativních rizik, které lze pak využít pro klasifikaci rizik. „Filtry" v podobě vážených faktorů nebo hranic skóre rizik, lze použít pro přizpůsobení klasifikace rizik cílům řízení či politiky.
Možné oblasti využití
Klasifikaci a filtrování rizik lze použít ke stanovení priorit výrobních míst z hlediska inspekcí/kontrol prováděných regulátory nebo průmyslem. Metody klasifikace rizik jsou zvláště užitečné v situacích, kdy je portfolio rizik a jejich základních důsledků, které je třeba řídit, diverzifikované a obtížně se porovnává pomocí jediného nástroje. Klasifikace rizik je užitečná tehdy, pokud vedení potřebuje vyhodnotit rizika posouzená kvalitativně i kvantitativně v rámci jedné organizační struktury.
I.9 Podpůrné statistické nástroje
Statistické nástroje mohou podporovat a usnadňovat QRM. Mohou umožnit efektivní posouzení dat, napomoci při stanovování významu datových souborů a podpořit spolehlivější rozhodování. Výčet některých hlavních statistických nástrojů používaných ve farmaceutickém průmyslu je zde:
(i) Regulační diagramy, např.:
- Přejímací regulační diagramy (viz ISO 7966)
- Regulační diagramy pro aritmetický průměr s výstražnými mezemi (viz ISO 7873
- Diagramy pro metodu kumulovaných součtů (viz ISO 7871)
- Shewhartovy regulační diagramy (viz ISO 8258)
- Vážený pohyblivý průměr
(ii) Navrhování experimentů (DOE)
(iii) Histogramy
(iv) Paretovy diagramy
(v) Analýza způsobilosti procesu
Příloha II: Možná uplatnění QRM
Cílem této přílohy je stanovit možná využití principů a nástrojů QRM ze strany průmyslu a regulátorů. Nicméně výběr konkrétních nástrojů řízení rizik zcela závisí na konkrétních skutečnostech a okolnostech.
Tyto příklady jsou předkládány pro ilustraci a slouží pouze jako návrhy možného využití QRM. Cílem této přílohy není vytvořit žádná nová očekávání nad rámec stávajících regulačních požadavků.
II.1 QRM jako součást integrovaného managementu jakosti
Dokumentace
Přezkoumání stávajícího výkladu a aplikace regulačních očekávání.
Stanovení vhodnosti a/nebo vývoj obsahové náplně SOP, pokynů apod.
Školení a vzdělávání
Stanovení vhodnosti zaškolení a následných školení podle vzdělání, zkušeností a pracovních návyků pracovníků a dále na základě pravidelného hodnocení předchozích školení (např. jejich efektivity).
Stanovení školení, zkušeností, kvalifikace a fyzických schopností, které umožňují pracovníkům provádět činnosti spolehlivě a bez nežádoucích vlivů na jakost přípravku.
Závady v jakosti
Vytvoření platformy pro stanovení, vyhodnocení a komunikaci potencionálního dopadu podezření na závadu jakosti, stížnosti, trendu, odchylky, šetření, výsledku nevyhovujícímu specifikacím apod.
Pomoc při komunikaci rizik a stanovení vhodných kroků pro řešení významných závad v jakosti společně s regulačními orgány (např. stahování z trhu).
Audity/inspekce
Stanovení frekvence a rozsahu auditů, interních i externích, při zohlednění takových faktorů, jako jsou:
- Stávající právní požadavky
- Celkový stav dodržování předpisů a historie společnosti či zařízení
- Robustnost činností voblasti QRM vdané společnosti
- Složitost závodu
- Složitost výrobního procesu
- Složitost přípravku a jeho léčebný význam
- Počet a významnost závad jakosti (např. stažení)
- Výsledky předchozích auditů/inspekcí
- Hlavní změny vbudovách, zařízení, procesech, klíčových pracovnících
- Zkušenosti svýrobou přípravku (např. frekvence, objemy, počet šarží)
- Výsledky zkoušek provedených vúředních kontrolních laboratořích
Periodické přezkoumání
Výběr, vyhodnocení a interpretace výsledků trendů dat v rámci přezkoumání jakosti přípravku.
Interpretace dat monitorování (např. pro podporu posouzení vhodnosti revalidace nebo změn v odběru vzorků).
Řízení změn/kontrola změn
Řízení změn na základě znalostí a informací shromážděných při farmaceutickém vývoji a výrobě.
Vyhodnocení dopadů změn na dostupnost konečného přípravku.
Vyhodnocení dopadů změn v zařízení, vybavení, materiálu, výrobním postupu nebo technologických transferech na jakost přípravku.
Stanovení vhodných kroků předcházejících implementaci změny, např. dodatečné testování, (re)kvalifikace, (re)validace nebo komunikace s regulátory.
Neustálé zlepšování
Usnadnění neustálého zlepšování procesů v rámci celého životního cyklu přípravku.
II.2 QRM jako součást regulačních činností
Činnosti inspekce a posuzování
Pomoc při přidělování zdrojů, včetně např. plánování a četnosti inspekcí a intenzitě inspekcí a posuzování (viz část „Audity" v příloze II.1).
Hodnocení významu např. závad v jakosti, potencionálních stažení a inspekčních nálezů.
Stanovení vhodnosti a typu poinspekčního regulačního následného sledování.
Vyhodnocení informací předkládaných průmyslem, včetně informací o farmaceutickém vývoji.
Vyhodnocení dopadů navrhovaných změn.
Identifikace rizik, o nichž je třeba informovat další inspektory a posuzovatele s cílem napomoci lepšímu pochopení možností kontroly rizik (např. parametrické propouštění, PAT (Process Analytical Technology).
II.3 QRM jako součást vývoje
Navržení kvalitního přípravku a jeho výrobního procesu tak, aby byla bez výkyvů zajištěna zamýšlená funkce přípravku (viz ICH Q8).
Zvýšení znalostí o funkci přípravku v široké šále materiálních atributů (např. distribuce velikosti částic, obsah vlhkosti, tokové vlastnosti), procesních možností a procesních parametrů.
Posouzení kritických atributů surovin, rozpouštědel, výchozích surovin účinné léčivé látky (API), účinných léčivých látek, pomocných látek nebo obalových materiálů.
Stanovení odpovídajících specifikací, identifikace kritických procesních parametrů a zavedení výrobních kontrol (např. využívání informací z vývojových farmaceutických studií týkajících se klinického významu nebo atributů jakosti či schopnosti kontrolovat je během zpracování).
Snížení variability jakostních atributů:
- snížení vad přípravku a materiálů,
- snížení výrobních vad.
Posouzení potřeby dalších studií (např. bioekvivalence, stability) vzhledem k navyšování objemu výroby a transferu technologií.
Využití konceptu „design space" (viz ICH Q8).
II.4 QRM pro zařízení, vybavení a inženýrské sítě
Návrh zařízení/vybavení
Stanovení vhodných zón při projektování budov a zařízení, např.:
- Pohyb materiálu a personálu
- Minimalizace kontaminace
- Deratizační opatření
- Prevence záměny
- Otevřené zařízení versus uzavřené zařízení
- Čisté prostory versus izolátorové technologie
- Vyhrazená či segregovaná zařízení/vybavení
Stanovení vhodných materiálů zařízení a kontejnerů přicházejících do styku s přípravky (např. výběr třídy nerezové oceli, těsnicích vložek, mazadel).
Stanovení vhodných inženýrských sítí (např. pára, plyny, zdroje elektřiny, stlačený vzduch, topení, vzduchotechnika, voda).
Stanovení vhodné preventivní údržby pro související vybavení (např. soupis nezbytných náhradních dílů).
Hygienické aspekty zařízení
Ochrana přípravku před nebezpečím plynoucím z prostředí, včetně chemických, mikrobiologických a fyzikálních nebezpečí (např. určení vhodného oděvu, hygienické otázky).
Ochrana prostředí (např. personál, potenciál křížové kontaminace) před nebezpečím souvisejícím s výrobou přípravku.
Kvalifikace zařízení/vybavení/inženýrských sítí
Stanovení rozsahu kvalifikace zařízení, budov a výrobního vybavení a/nebo laboratorních přístrojů (včetně patřičných metod kalibrace).
Čištění zařízení a kontrola prostředí
Rozlišení programů čištění na základě účelu použití (např. víceúčelové zařízení oproti jednoúčelovému, šarže oproti průběžné výrobě).
Stanovení přijatelných (specifikovaných) limitů validace čištění.
Kalibrace/preventivní údržba
Nastavení vhodných harmonogramů kalibrace a údržby.
Počítačové systémy a počítačově řízené vybavení
Výběr designu počítačového hardwaru a softwaru (např. modulární, strukturovaný, tolerance chyb).
Stanovení rozsahu validace, např.:
- Identifikace kritických parametrů výkonnosti
- Výběr požadavků a designu
- Revize kódu
- Rozsah testování a testovací metody
- Spolehlivost elektronických záznamů a podpisů
II.5 QRM jako součást skladového hospodářství
Posouzení a vyhodnocení dodavatelů a smluvních výrobců
Zajištění komplexního vyhodnocení dodavatelů a smluvních výrobců (např. audity, dodavatelské smlouvy o jakosti).
Výchozí suroviny
Posouzení rozdílů a možných rizik pro jakost souvisejících s variabilitou výchozích materiálů (např. stáří, způsob syntézy).
Použití materiálů
Stanovení toho, zda je vhodné používat materiál v karanténě (např. pro další interní zpracování).
Stanovení vhodnosti opakovaného zpracování, přepracování, použití vráceného zboží.
Podmínky skladování, distribuce a logistika
Posouzení přiměřenosti opatření k zajištění zachování vhodných podmínek skladování a přepravy (např. teplota, vlhkost, návrh kontejneru).
Stanovení vlivu nesrovnalostí v podmínkách skladování či přepravy na jakost přípravku (např. řízení chladového řetězce) ve spojitosti s dalšími pokyny ICH.
Zachování infrastruktury (např. kapacity pro zajištění vhodných podmínek přepravy, dočasného skladování, manipulace s nebezpečnými materiály a kontrolovanými látkami, celní odbavení).
Poskytnutí informací pro zajištění dostupnosti léčiv (např. klasifikace rizik pro dodavatelské řetězce).
II.6 QRM jako součást výroby
Validace
Stanovení rozsahu činností verifikace, kvalifikace a validace (např. analytické metody, procesy, vybavení a metody čištění).
Stanovení rozsahu následných činností (např. odběr vzorků, monitorování a revalidace).
Odlišení kritických a nekritických kroků v procesech pro účely snazšího navržení validační studie.
Mezioperační a průběžné vzorkování a zkoušení
Vyhodnocení frekvence a rozsahu mezioperačního kontrolního zkoušení (např. za účelem zdůvodnění redukovaného zkoušení v podmínkách doložené kontroly).
Vyhodnocení a odůvodnění aplikace PAT (Process Analytical Technologies) společně s parametrickým propouštěním a propouštěním v reálném čase.
Plánování výroby
Stanovení odpovídajícího plánování výroby (např. vyhrazených, kampaňových a souběžných sekvencí výrobního procesu).
II.7 QRM jako součást laboratorní kontroly a stabilitních studií
Výsledky nevyhovující specifikacím
Stanovení potencionálních původních příčin a nápravných opatření během šetření výsledků nevyhovujících specifikacím.
Lhůty reatestace/data ukončení použitelnosti
Vyhodnocení adekvátnosti skladování a zkoušení meziproduktů, pomocných látek a výchozích surovin.
II.8 QRM jako součást balení a značení
Návrh obalů
Navržení sekundárního balení jakožto ochrany primárně zabaleného přípravku (např. pro zajištění autentičnosti přípravku, čitelnosti údajů na obalu).
Výběr systému uzavření kontejneru
Stanovení kritických parametrů systému uzavření kontejneru.
Kontroly značení
Navržení postupů kontroly značení na základě možnosti vzniku záměn vztahujících se na značení různých přípravků, včetně různých verzí téhož značení.





 cz
cz